B M B 400
Part Four: Gene
Regulation
Section VI = Chapter 20
REGULATION BY CHANGES IN CHROMATIN STRUCTURE
Review of nucleosome and chromatin structure
Nucleosome composition
Nucleosomes are the repeating subunit of chromatin.
Nucleosomes are composed of a nucleosome core, histone H1 (in higher eukaryotes) and variable length linker DNA (0-50bp).
The nucleosome core contains an octamer of 2 each of the core histones (H2A, H2B, H3 and H4) and 146 bp of DNA wrapped 1.75 turns (Fig. 4.6.1).
Core histones are small basic
proteins (11-14 kDa) that contain a central structure histone-fold domain and
N-terminal and C-terminal
extensions.
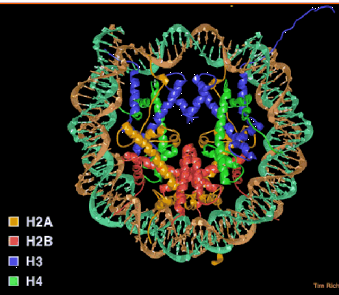
Figure 4.6.1. Nucleosome core particle. A “top” view derived from the three-dimensional structure deduced in T. Richmond’s laboratory.
Histone interactions in the nucleosome
Core histones dimerize through their histone fold motifs generating H3/H4 dimers and H2A H2B dimers (Fig. 4.6.2.).
Two H3/H4 dimers associate to form a tetramer, which binds DNA.
Two H2A/H2B dimers associate with the tetramer to form the histone octamer.
At physiological salt the octamer is not stable unless bound to DNA and dissociates into the H3/H4 tetramer and two H2A/H2B dimers.
Each histone pair bends approximately 30bp of DNA around the histone octamer.
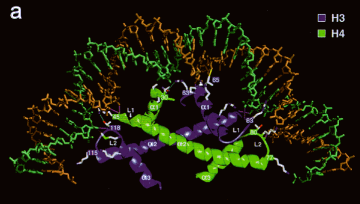
Figure 4.6.2. An H3-H4 dimer bound to DNA.
Chromatin higher order structure
Arrays of nucleosomes condense into higher order chromatin fibers (Fig. 4.6.3.).
Despite over 2 decades of investigation the structure of the “30nm” chromatin fiber is not known.
This may be due to irregularity or instability of the structure.
This level of structure has been implicated in mechanisms of chromatin repression; thus, the lack of structural information at this level is particularly troublesome

Figure 4.6.3. Solenoid model for the 30 nm chromatin fiber.
Different states (degree of compaction) of chromatin correlate with gene activity.
Chromatin, not naked DNA, is the substrate for transcription, replication, recombination, repair and condensation during mitosis and meiosis. Thus the extent of compaction of the chromatin in the different states will affect the ability of transcription factors, polymerases, repair enzymes, and the recombination machinery to access this substrate. More open, accessible chromatin is associated with greater transcriptional activity.
Condensed chromatin is transcriptionally inactive (usually)
Heterochromatin is defined cytologically as the densely staining, localized material containing DNA in the interphase nucleus (Fig. 4.6.4.). Other DNA-containing material stains more lightly, diffusely across the interphase nucleus; it is called euchromatin. Higher resolution microscopy shows that heterochromatin contains thicker fibers of chromatin, and hence is more compact than euchromatin. Some, and perhaps much, of the DNA in heterochromatin is highly repeated. For instance, centromeres (and the regions around them) and telomeres are composed of short DNA sequences repeated many times. These tend to be in heterochromatin. Also, rRNA genes are highly repeated and are in heterochromatin.
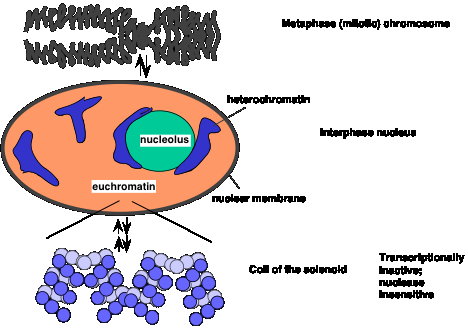
Figure 4.6.4. Compact chromatin in metaphase and interphase, and shifts to more open euchromatin.
Many of the DNA sequences in heterochromatin are not transcribed. The rRNA genes are a notable exception; they are abundantly transcribed, but most heterochromatic DNA is not.
Several lines of evidence support the association of tightly folded, compact heterochromatin is associated with gene silencing. One is the phenomenon of position effect variegation (PEV). This refers to change in the level of gene expression as a function of chromosomal position (position effect). The phenotype varies among cells in a tissue or population; hence it is a variegating phenotype.
A classic example of PEV results from a chromosomal inversion affecting eye color in flies. Inversions of a segment of a chromosome that places the w+ gene close to constitutive heterochromatin lead to position effect variegation (Fig. 4.6.5).
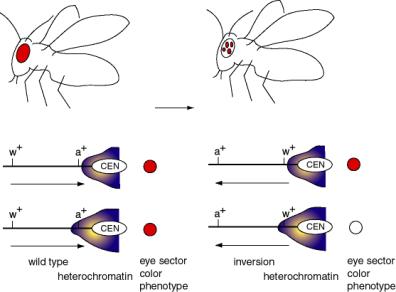
Figure 4.6.5. Position effect variegation caused by differential expansion of pericentromeric heterochromatin.
The wild-type w+ gene, in its normal chromosomal position, causes red eyes in Drosophila melanogaster. Mutant alleles can have no red color (i.e. the classic w-, the first Drosophila mutant, discovered by the Mrs. T.H. Morgan) or many modifications of red (apricot, cinnabar, etc.).
Chromosomal inversions have been isolated that generate a variegated eye-color - patches of red on a white background. In these cases, the wild-type w+ gene is still present, but it is now close to or within the heterochromatic region close to the centromere (because of the inversion).
There is not a precise boundary to the heterochromatin, so in some clones of cells (in a particular segment of the eye) the heterochromatin encompasses the w+ gene, turning it off, and giving a white color. In other clones of cells, the heterochromatin does not cover the w+ gene, and these segments of the eyes form the red patches. The variegation derives from clonal differences in the extent of heterochromatin.
The main point is that a wild-type w+ gene can either be expressed or not, depending on the type of chromatin it is in.
Less condensed chromatin is associated with transcriptional activity
(active chromatin)
Review:
Wolfe, A. (1994) TIBS 19:231-267.
The explanation for the activity of the translocated w gene in some cells is that it is not condensed into heterochromatin in those cases. Several lines of evidence support an association between more open (less condensed) chromatin and gene activity. The basic idea is that active chromatin is more "open" (accessible to proteins and reagents) than is bulk chromatin.
Cells that are actively expressing their genes have larger nuclei than do transcriptionally quiescent cells.
Treatment of Drosophila cells with ecdysone (a steroid hormone) generates visible "puffs" at defined loci on the polytene chromosomes - the loci with ecdysone-inducible genes (Fig. 4.6.6). In these puffs, the chromatin extends out and is actively transcribed. These are the sites of incorporation of labeled ribonucleoside triphosphates into RNA, as demonstrated by autoradiography.
Heat shock treatment of Drosophila also generates puffs, but at different loci - those with the heat shock genes. The heat shock puffs are bounded by specialized chromatin structures called scs and scs'.
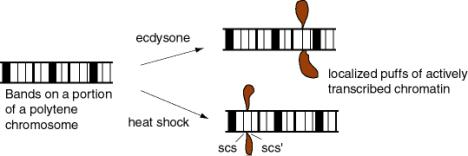
Figure 4.6.6.
Although this association of active transcription with more accessible chromatin is well established, the structures of the more accessible and less accessible chromatin have not been clearly defined (Fig. 4.6.7).
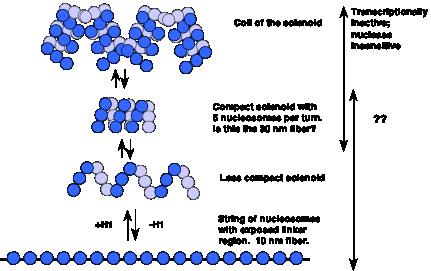
Figure 4.6.7. More open chromatin can be transcriptionally active
Biochemical investigation of different states of chromatin and gene activity in cells
Sensitivity of chromatin to nucleases
A seminal observation in the correlation of gene activity with more accessible chromatin was the demonstration that transcriptionally active genes are found in chromatin that is more sensitive to DNases. Weintraub and Groudine showed in 1976 that the overall sensitivity of a gene to DNase I is increased about 3 to 10 fold over that of DNA in bulk chromatin, but only in tissues expressing the gene (Fig. 4.6.8). Subsequent studies have shown this correlation for many genes in many tissues, but it is not seen in every case. Some genes are in accessible chromatin whether they are expressed or not. The reasons for these differences are being studied.
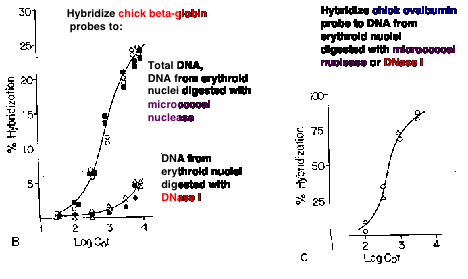
Figure 4.6.8. DNase I digestion of nuclei reduces the concentration of actively transcribed DNA. Adapted from Stalder et al. (1980) Cell 20:451-460.
The basic experimental approach was to measure the sensitivity of particular sequences to nuclease digestion in nuclei from expressing and nonexpressing tissues (Fig. 4.6.8). For example, nuclei from chicken erythroid cells (avian red blood cells retain their nuclei, in contrast to mammals) and liver cells were digested separately with DNase I. Sufficient nuclease was added so that sensitive regions would be cut but the bulk of the DNA in chromatin was only lightly digested. Chromosomal proteins were then removed (proteinase K followed by phenol extraction) leaving purified DNA. The partially digested nuclear DNA was denatured and annealed to labeled gene-specific hybridization probes, and the appearance of the labeled probe in duplex with the nuclear DNA was monitored as a function of Cot (concentration of DNA ´ time - recall this from Part One of the course). DNA from partially digested liver nuclei annealed with the globin gene probe at a much lower Cot than did DNA from partially digested erythroid nuclei. This shows that the amount of globin gene DNA in erythroid nuclei is substantially reduced by the DNase I treatment, i.e. the globin gene is sensitive to DNase I in a cell that is expressing it. {To put a finer touch on it, the erythrocytes are descended from cells that were actively expressing globin genes. In this particular case, formerly expressed genes retain their DNase I sensitivity.}
An important negative control is the annealing to a labeled ovalbumin gene probe, a gene that is not expressed in either liver or red cells (only oviduct). In this case, the DNA from partially digested nuclei from both tissues annealed with the same kinetics to the ovalbumin probe. Thus there is no gross over-digestion of the erythroid nuclei, and it is clear the globin gene is much less sensitive to nucleases in nonexpressing tissues.
Mapping the extent of the region around the gene that is accessible
The basic strategy is similar to that used above, but the nuclear DNA is monitored as a function of [DNase I], hybridization probes from outside the gene are used, and a blot-hybridization assay is employed (Fig. 4.6.9). After obtaining the DNA from nuclei digested to increasing extents with DNase I, the DNA is digested to completion with restriction endonucleases, separated by size on an agarose gel, blotted to a membrane like nylon and hybridized with a radioactive probe from within the gene or from regions flanking the gene. Probes from within and immediately flanking the gene show a progressive loss of signal as the [DNase I] is increased in the initial digestion, hence the name "fade-out" experiments for these assays. Further away from the gene, once one is outside the open domain, the signal from the restriction fragments does not decline any faster than the negative control. The boundaries of the open domain lie outside the fragments that show sensitivity but inside the fragments that show insensitivity.
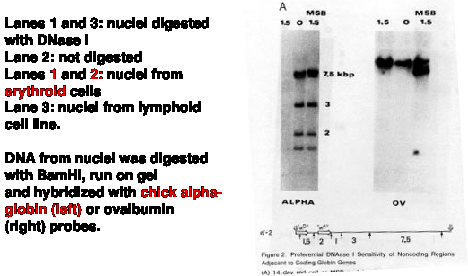
Figure 4.6.9. DNase I digestion of nuclei preferentially cuts restriction endonuclease fragments containing actively transcribed DNA. Adapted from Stalder et al. (1980) Cell 20:451-460, Fig. 2,
In the case of the human b-like globin gene cluster (see below), the region for insensitivity begins over 60 kb 5' to the b-globin gene and over 100 kb 3' to it. In other cases, e.g. chicken lysozyme gene, the entire domain is about 20 kb in size and has a single gene.
The structural basis for the increased sensitivity to digestion by DNase I in cells is not firmly established. It is often interpreted as being the result of unfolding in higher order structure. One possibility is that DNA that is sensitive over a broad region is in the 10 nm fiber (a linear string of nucleosomes), whereas insensitive regions may be in a 30 nm fiber, which is thought to be a solenoid of nucleosomes. However, some genes in the 30 nm fiber may be active, and inactivation may correspond to a higher order compaction, or assembly of a silencing structure.
The extended regions of general DNase sensitivity are thought to define a functional domain in chromatin. It may correspond to a large loop of chromatin (e.g. 100 kb or more) (Fig. 4.6.10).
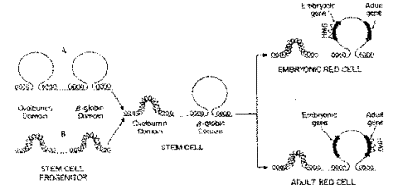
Figure 4.6.10. Regions of general DNase sensitivity may correspond to "lampbrush” chromosome-like loops or domains. Adapted from Stalder et al., 1980, Cell 20:451
DNase hypersensitive sites
Specific, short regions (usually about 100 to 200 bp) are about 100 times more sensitive that bulk DNA in nuclei. Because DNase I cuts frequently in this short region, it generates a double-stranded break at this hypersensitive site (abbreviated HS). This produces a new band on a genomic blot-hybridization assay (Fig. 4.6.11).
The technique employed, called "indirect end labeling" is a modification of the "fade-out" experiment described in Fig. 4.6.9 above, and it is used to detect HSs. As in the previous assays, nuclei are digested with increasing amounts of DNase I, DNA is purified and cleaved with a restriction endonuclease and the region of interest analyzed by genomic blot-hybridization (Southern blot). By using a radioactive probe from one end of the restriction fragment that is being detected on the genomic blot-hybridization assay (instead of the larger probes used in the previous assays), one can resolve the new fragments generated by cleavage by DNase I at a HS. The size of the new fragment tells you the position of the HS. For example, a new 5 kb fragment would mean that a HS is located 5 kb away from the restriction endonuclease cleavage site that is closest to the probe used in the assay.
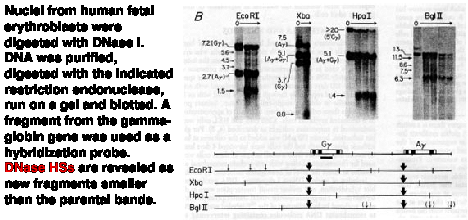
Figure 4.6.11. Indirect end-labeling assay maps DNase hypersensitive sites. This example uses Indirect end-labeling to see DNase HSs in gamma globin genes. Adapted from Groudine et al. (1983) PNAS 80:7551-7555.
This approach can reveal multiple hypersensitive sites (Fig. 4.6.12) as well as single site.
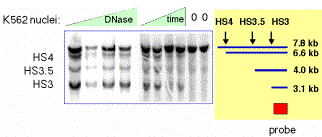
Figure 4.6.12. Example of results from an indirect end labeling assay. This experiment maps three DNase HSs in the human beta-globin locus control region (see Section E of this chapter0. Data from H. Petrykowska.
General properties of DNase HSs in chromatin
(1) HSs are free of nucleosomes, or the nucleosomes are highly disrupted. E.g. the SV40 control region is a HS, and visualization in the EM shows that SV40 minichromosomes do not have nucleosomes in this region.
(2) DNA sequences that are in HSs in chromatin are frequently involved in gene regulation. Examples are promoters, enhancers, silencers and LCRs. Matrix and scaffold attachment regions (MARs and SARs) are also hypersensitive to DNase I.
(3) Investigation of the HSs shows that they have multiple sites for binding transcription factors (as expected for promoters, enhancers, silencers, etc.) or other regulatory or structural proteins (e.g. MARs binding topoisomerase II).
(4) The basic idea is that the DNA can be occupied by specific binding factors (when the gene is being transcribed) or it can be wrapped into nucleosomes. In most (but not all) cases these are mutually exclusive options. The DNA is not hypersensitive to DNase I cleavage when it is in nucleosomes. The coverage of the DNA by the transcription factors is not complete and still allows cleavage by DNase I between the bound factors.
(5) The DNase HSs are landmarks for gene regulatory sequences.
Detailed analysis of active chromatin in a specific locus
Many aspects of the chromatin structure have been determined for the active beta-like globin genes in chicken erythroid cells. These are summarized in Fig. 4.6.13.
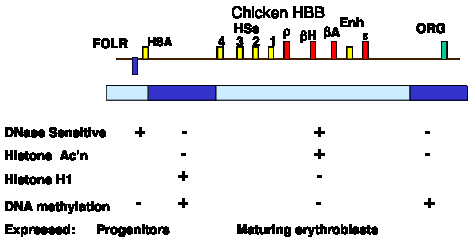
Figure 4.6.13. Biochemically defined domain can correspond to a set of coordinately expressed genes. Only developmentally stable DNase HSs are shown. The promoter for each gene also acquires a HS at the stage of development at which it is expressed.
1. A discrete region is accessible to nucleases (e.g. DNase I)
2. Demethylation of DNA
Actively expressed DNA has reduced levels of 5-methylcytosine at CpG dinucleotides. A very clear example of this is X-chromosome inactivation - several loci on the inactive X are highly methylated, whereas the alleles on the active X are much less methylated.
3. Depletion of histone H1
Since H1 seems to play a role in stabilizing the 30 nm fiber, then removal of H1 may aid the transition to the more open 10 nm fiber.
4. Acetylation
of core histones
All four core histones can be acetylated on lysines in their N-terminal tails, outside the hydrophobic core that constitutes the histone fold in the tertiary structure (histone structure was covered in Part One of the course). This acetylation is highly dynamic, with acetyl groups being added and taken off every few seconds. However, the core histones in chromatin containing actively transcribed genes are more highly acetylated than are the histones in the rest of the nucleus. Thus in active chromatin, the rate at which acetyl groups are added (by histone acetyl transferases, see below) exceeds the rate at which they are removed (by histone deacetylases).
The recent identification of specific histone acetyl transferases and the recognition that they comprise particular subunits of transcriptional co-activators have confirmed the intimate relationship between histone acetylation in chromatin and activation of gene expression. Thus further analysis of the mechanistic details of how this histone modification leads to changes in rates of transcription or other steps in gene expression is now being pursued intensively.
5. Ubiquitination of H2A
Ubiquitin is a 76 amino acid protein required for ATP-dependent, nonlysosomal, intracellular protein degradation. It is also found on some histones, e.g. a small fraction of H2A is covalently attached to ubiquitin (in fact this was how ubiquitin was discovered). The ubiquitination of H2A is not thought to be a signal for proteolysis (histones, like DNA, basically do not turn over during the life of a cell) but may be a signal to induce chromatin remodeling.
6. Nonhistone proteins HMG14 and HMG17
These members of the high mobility group of nonhistone chromosomal proteins are preferentially associated with active chromatin.
7. Nucleosome phasing
If all the copies of a gene in a population of cells (e.g. in a given tissue) have the same sequences in the nucleosome core and the same sequences in the linkers between the cores, we say those nucleosomes are in phase. This can arise, e.g., by having a strong preference for initiating nucleosome assembly at a particular short sequence. In those cases where nucleosomes are in phase, they can bring the binding sites for transcription factors into the proper array and orientation for the factors to bind. An example is the promoter/enhancer for MMTV.
9. Domain
boundaries
a. Only a few domain boundaries are well characterized.
boundaries of the chromosomal domain for the chicken lysozyme gene
5' end of the chromosomal domain for the chicken b-globin gene
b. They may play a passive role, protecting from the effects of adjacent sequences. That is, they insulate from position effects.
c. They are close to MARs in the case of the chicken lysozyme gene. However, not every MAR is a domain boundary.
Insulators are operationally defined by their ability to block activation of promoter by an enhancer (Fig. 4.6.14). The 5’ HS4 from the chicken HBB locus is an insulator, and also marks a boundary between accessible and inaccessible chromatin.
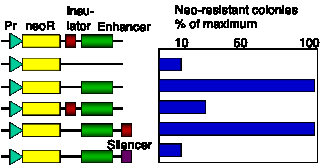
Figure 4.6.14. Assay for chromatin insulators. Results of a colony formation assay for HS4 from chick HBB complex are shown.
Opening of a chromatin domain is distinct from transcriptional activation
Some distal control elements have been implicated in chromatin-mediated regulation
Key regulatory sequences can be distal to genes, such as the locus control region (LCR) regulating the beta-like globin gene complex (HBBC) in mammals (Fig. 4.6.15).
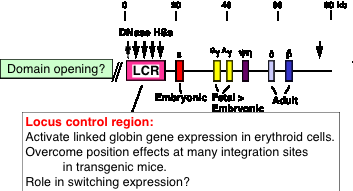
Figure 4.6.15. Human b-globin gene cluster
The ability of the HBBC LCR to allow expression of the beta-like globin genes at many different chromosomal positions indicates that it confers an ability to overcome negative position effects (Fig. 4.6.16). This has been interpreted as having an activity that will open a chromatin domain.

Figure 4.6.16. HBBC LCR will activate expression at many chromosomal locations
Examination of domain opening and gene activation
The proposed connection between enhancement of gene expression and opening a chromatin domain are actively being investigated. Experiments altering the LCR within the context of the entire chromosome show that different sequences are needed for domain opening and gene activation (Fig. 4.6.17). At this locus, the LCR is needed for transcriptional activation, not opening a domain so that it is DNase sensitive.
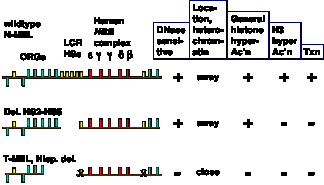
Figure 4.6.17. Domain opening and gene activation are separable events. Adapted from Reik et al. (1988) Mol. Cell. Biol. 18:5992-6000 and Schübeler et al. (2000) Genes & Devel. 14:940- 950.
The opening of a chromatin domain is associated with the movement of the locus within the interphase nucleus to a region without heterochromatin, as shown by in situ hybridization analysis with gene specific probes (Fig. 4.6.18). Thus more closed chromatin is physically associated with heterochromatin. Movement away from heterochromatin correlates with domain opening, but it does not necessarily lead to gene activation. Movement away from heterochromatin (presumably into euchromatin) may be a prerequisite for activation.
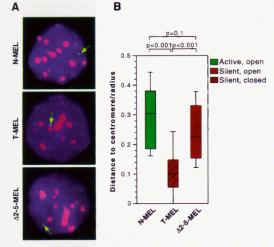
Figure 4.6.18. Domain opening is associated with movement to non-heterochromatic regions.
Proposed sequence for gene activation
1. Open a chromatin domain
Relocate away from pericentromeric heterochromatin
Establish a locus-wide open chromatin configuration
General histone hyperacetylation
DNase I sensitivity
2. Activate transcription
Local hyperacetylation of histone H3
Promoter activation to initiate and elongate transcription
Summary of cis-regulatory elements that act in chromatin
Generate an open, accessible chromatin structure
Can extend over about hundreds of kb
Can be tissue specific
Enhance expression of individual genes
Can be tissue specific
Can function at specific stages of development.
Insulate genes from position
effects.
Enhancer blocking assay
How is the structure of chromatin modified in cells to change transcriptional activity?
Competition vs. Replacement models for how transcription factors occupy their binding sites on a chromatin template.
a. The competition model requires DNA replication to expose the factor binding sites. When nucleosomal DNA is replicated, half of the DNA is free of nucleosomes, at least transiently, prior to the formation of more nucleosomes. This gives the opportunity for transcription factors to bind - they just have to do it before more nucleosomes assemble. Thus there is competition between nucleosome formation and factor binding.
b. An alternative model is that the transcription factors replace the nucleosomes in an active process. Some mechanism may disrupt or dissociate the nucleosomes, allowing the factors to bind. DNA replication is not a pre-requisite for replacement.
c. There are examples that conform to each of these models, i.e. either may apply to a given gene.
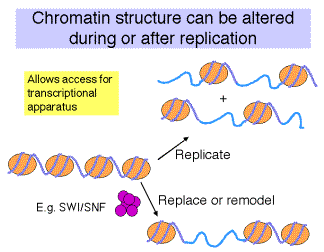
Fig. 4.6.19
The conformation of chromatin can be altered in vitro
This can be seen in the different states of chromatin in the EM views in Fig. 4.6.20. More condensed chromatin can be induced by increasing the salt concentration of the amount of H1 histone.
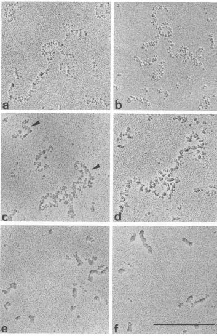
Figure 4.6.20.
Enzymatic activities implicated in chromatin remodeling and gene activation
As discussed before, transcriptional activation of genes is associated with the binding of activator proteins to promoters and enhancers. Chromatin-mediated activation is thought to occur by stimulating the sequence-specific binding of activators in chromatin. At least four different classes of activities have been identified that aid binding of activators.
1. Cooperative binding of multiple factors.
2. The presence of histone chaperone proteins, which can compete H2A-H2B dimers from the nucleosome.
3. Acetylation of the N-terminal tails of the histones.
4. Nucleosome disruption by ATP-dependent remodeling complexes.
These will be considered in the subsequent sections.
Binding of transcription factors and effects of chaperones
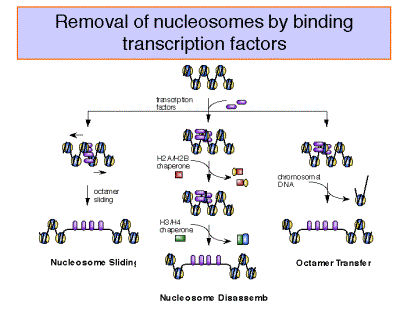
Figure 4.6.21.
"Remodeling" ATPases:
These large, multisubunit complexes usually have one component with an ATPase "domain" and/or activity, some of which match a helicase family. One idea is that these ATPases destabilize the nucleosome core, allowing H2A-H2B dimers to dissociate (and bind to chaperones like nucleoplasmin) and promoting binding of transcription factors.
Recent studies show that the action of the remodeling ATPase results in a stably altered nucleosome (Fig. 4.6.22), but the exact nature of the alterations is still being investigated. The full complement of histones remains on the remodeled nucleosome, which is more accessible to transcription factors as well as nucleases. The enzymes can shift the altered nucleosome back to a standard nucleosome in an ATP-dependent process, showing that the alterations are reversible (Schnitzler et al. 1998, Cell 94:17-27; Lorch et al. 1998, Cell 94: 29-34).
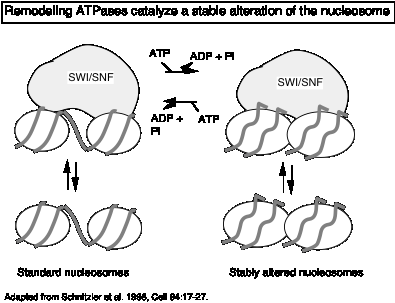
Figure 4.6.22.
Examples of these remodeling ATPases include yeast SWI/SNF, its mammalian homolog Brahma and yeast RSC. The SWI/SNF complex is a very large complex containing about 11 different proteins. Each of these components was identified genetically as being required for the activation of a large number of genes in yeast (but not all genes). They were initially discovered as genes required for expression of the gene encoding HO endonuclease, which plays as key role in mating type switching (hence the SWI designation), and the gene for invertase (or sucrase - it splits sucrose into glucose and fructose). Mutants in these genes cannot utilize sucrose as a carbon source (sucrose nonfermenting or snf). All 5 proteins form a large complex. SWI/SNF is needed for the activation of a subset of inducible genes, whereas RSC is required for viability.
Some suppressors of swi or snf mutants turned out to be mutations in genes encoding histones. This indicated that the SWI/SNF complex could interact with chromatin to activate the target genes; recent biochemical studies show this very clearly (see above citations and references therein).
In vitro data show that the SWI/SNF complex will facilitate the binding of a transcriptional activator (a modified GAL4 protein) to nucleosomal cores in an ATP-dependent manner.
The SWI/SNF complex is the prototype cellular machine that alters, or remodels nucleosomes to allow easier access to transcription factors and in some way activation gene expression.
The mammalian homolog is hSWI/SNF. The ATPase is BRG1, which is related to the Drosophila Brahma protein.
Some remodeling ATPases may be specific to certain classes of genes. The X-linked ATR locus may be an example of this.
Histone acetyl transferases:
Histones are covalently modified during replication, gene activation and gene repression. Often these modifications are in the N-terminal tails, which protrude from the nucleosomal core particle. The types and sites of covalent modification for H3 and H4 are shown in Fig. 4.6.23.
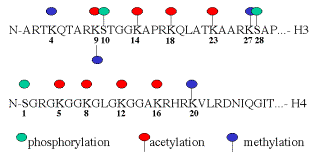
Fig. 4.6.23. Sites and types of covalent modification of histone tails.
A major modification of histones is acetylation. As shown in Fig. 4.5.23, multiple lysine residues are targets for acetylation. Not all sites are acetylated in any one cellular process. Acetylation of some lysines is associated with replication, whereas acetylation of others is associated with gene activation. Deacetylation is associated with repression or silencing of genes (Fig. 4.6.24).
The major roles being studies for histone acetylation in gene activation are:
a) Increase the access of transcription factors to DNA in nucleosomes.
b) Decondensation of higher order chromatin structures (e.g. 30 nm fibers).
c) Serve as markers for the binding of nonhistone proteins. An example is bromodomain proteins, which are components of the nucleosome remodeling complexes.
The basic biochemical reaction of histone acetylation is the addition of an acetyl group to the e-amino group of lysine (Fig. 4.6.24). This reaction uses acetyl CoA as the donor of the acetate. The result of this reaction is a loss of one positive charge on the histone by one for every acetate that is added to a lysine.
Almost all the histones are acetylated. Histones H3 and H4, which make up the tetramer in the center of the nucleosome, can be highly acetylated (four or more acetates per histone).
Many of the acetylation sites are on the N-terminal tails that are outside the nucleosome core. Acetylation may alter the interactions between nucleosomes to allow some access to transcription factors.

Fig. 4.6.24.
The enzymes that add acetyl groups to the lysines of histones are called histone acetyl transferases, or HATs (Fig. 4.6.25). Recent biochemical and genetic evidence strongly supports a role for histone acetylation in activation of gene expression from chromatin templates, but much remains to be established about the mechanism.
The HATs are large, multisubunit complexes that will transfer acetyl groups from acetylCoA to the e-amino groups of lysines on histones in nucleosomes (Fig. 4.6.25). Several are recognized to date. Two prominent ones contain Gcn5p and Ada2, and one of these contains Spt proteins, which are thought to be required for TBP function. These "SAGA complexes" thus are adapters (needed for transcriptional activation), which appear to be very similar to "co-activators".
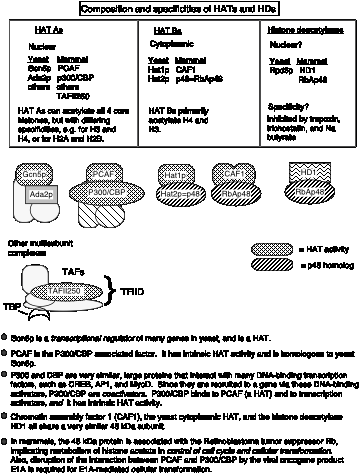
Figure 4.6.25. Different HAT complexes are used in chromatin assembly and modification. Histone deacetylation is associated with silencing.
Several lines of evidence show that nuclear HATs function as coactivators. They work together with other transcription factors for activation of many genes. Much of this evidence is derived from analysis of the components of the multisubunit HAT complexes in yeast and humans. Some of the key components are shown schematically in Fig. 4.6.26 and are listed in Table 4.6.1.
a) Some transcriptional activators are components of HAT complexes. One of the first examples was the protein Gcn5p from yeast. It had been previously characterized genetically as a transcriptional activator. When some HAT complexes were isolated, Gcn5p was found to be one of the subunits. Indeed, it has the catalytic acetyl transferase activity. In mammalian cells, a protein called PCAF (P300/CBP associated factor) is a HAT and is homologous to the yeast Gcn5p.
The proteins P300 and CBP (CREB-binding protein) are similar proteins that bind to a number of transcriptional activators (in addition to CREB, which binds to cAMP response elements, examples include MyoD and AP1). These large proteins are needed for activation by these factors. P300 and CBP have been shown to have HAT activity. In addition, they bind to another HAT, PCAF.
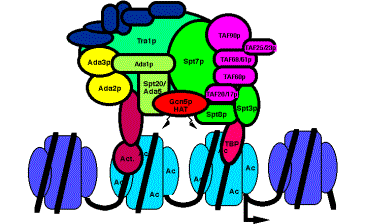
Figure 4.6.26. The yeast HAT complex called SAGA shown interacting with chromatin.
b) Proteins required for the function of some activators are components of HAT complexes. The Ada proteins were discovered as the products of genes that when mutated prevented a function of some transcriptional activators in yeast. They have been termed transcriptional adapters. Several Ada proteins are components of purified HAT complexes.
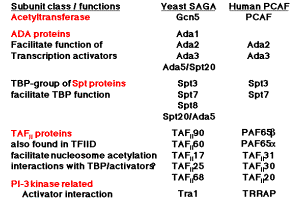
c) Proteins that interact intimately with TBP are also components of HAT complexes. Recent studies show that a subset of the TAFIIs are integral components of the SAGA (yeast) and PCAF (human) complex and are required for nucleosome acetylation and transcriptional stimulation (Grant et al. 1998, Cell 94: 45-53; Ogrysko et al. 1998, Cell 94: 35-44). The SPT proteins were shown genetically to regulate the function of TBP. Several of these are found in HATs in yeast and human (Fig. 4.6.26, Table 4.6.1).
The activator Gcn5p, the Ada transcriptional adapters, and the Spt proteins regulating TBP were discovered independently by different genetic assays. The biochemical purification of HAT complexes and identification of their subunits showed that these genetically distinguishable proteins are working together in a common complex. This complex was termed SAGA for the Spt proteins, Ada adapters, and Gcn5p components. This complex has the ability to catalyze acetylation of histones within nucleosome cores, and it is likely that this activity is a key part of the several functions of this complex in the cell.
Table 4.6.1. The high conservation in subunit composition of HAT complexes between yeast and human argues for a central role in transcription regulation.
d) Nucleosomal templates acetylated by purified HATs are more permissive for activated transcription in vitro. When a DNA containing a transcription unit is assembled into chromatin, it is transcribed in vitro much less efficiently than when it is free of histones; this is a nonspecific nucleosomal repression of transcription. Some transcriptional activators can boost transcription from such nucleosomal templates, but they require co-activators for this process. Many different proteins function in this assay, including TFIID (TBP plus TAFs) and P300/CBP. Recent studies show that reaction of a nucleosomal template with a purified HAT complex (such as SAGA) and acetyl CoA produces a template on which transcriptional activators are highly effective. This is a direct demonstration of co-activator function in vitro. Coupled with the extensive genetic evidence on the roles of the components of HATs, the case is strong for a role of HATs in coactivation in vivo (Fig. 4.6.27).
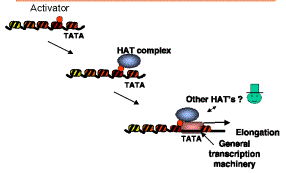
Figure 4.6.27. Model for HATs as co-activators.
Multiple nuclear HATs are found in yeast and in other species (Table 4.6.2). They are all large with many subunits. By comparison, their substrate, which is the nucleosome, is 0.2 MDa in mass. They have different substrate specificities. Some acetylated H3 preferentially, others acetylate H4. The reason for the diversity of HATs is a matter of current study.
Table 4.6.2. The four major nuclear HAT complexes in yeast
|
Complex |
Mass (MDa=megadaltons) |
|
|
SAGA |
1.8 |
|
|
NuA4 |
1.4 |
|
|
ADA |
0.8 |
|
|
NuA3 |
0.5 |
|
Histone deacetylases
These are implicated in chromatin-mediated repression (Fig. 4.6.28).
Methylation of DNA, followed by binding of proteins that recognize methylated DNA, can recruit histone deacetylases (HDACs). This is one mechanism of repression by methylation of DNA (Fig. 4.6.29).
![]()
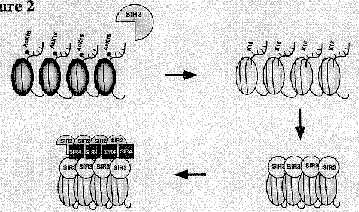
Figure 4.6.28. Repression by deacetylation of histones.
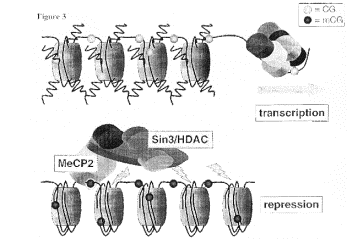
Figure 4.6.29. Methylated DNA can recruit HDACs.
Nucleosome remodeling and histone acetylation in nucleosomes are linked
This conclusion is an extrapolation from genetic evidence showing that the nucleosome remodeling activity of SWI/SNF and the acetylation of nucleosomes by SAGA are connected. In particular, some genes require both complexes for activation. Other genes require only one or the other complex, or neither. However, in these cases, their activation may utilize different ATP-dependent complexes and/or HATs.
One of the best-studied examples is that of the gene encoding the HO endonuclease in yeast. It requires both SWI/SNF and SAGA for activation, and they act in a particular order. The order of recruitment of factors to the promoter of the HO endonuclease gene is:
1) SWI5 activator
2) SWI/SNF nucleosome remodeling complex
3) SAGA histone acetyl transferase complex
4) SBF activator
5) General transcription factors
Ref: Cosma, Tanaka and Nasmyth (1999) Cell 97: 299-311.
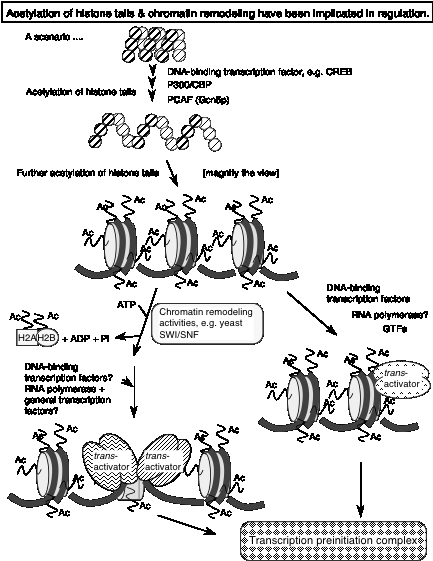
Fig. 4.6.30
Questions on Chapter 20. Regulation by changes in chromatin structure
Use the following information to answer the next two questions.
DNase hypersensitive sites around a gene were mapped by treating nuclei from cells that express that gene with increasing amounts of DNaseI. The partially digested DNA was isolated, cut to completion with a restriction enzyme, and analyzed by Southern blot-hybridization using a radioactive probe that is located 3' to the gene. Cleavage of genomic DNA with the restriction enzyme generates an 8 kb fragment that contains the gene, and the probe for the blot hybridization is located at the right end of the fragment (left to right defined as the direction of transcription of the gene). The results of this indirect end-labeling assay shows a gradual fade-out of the 8 kb fragment with increasing [DNaseI], and the appearance of a new band at 6 kb with DNaseI treatment.

20.1 Where is the DNase I hypersensitive site?
20.2 If the start site for transcription is 5 kb from the right end of the restriction fragment, what is a likely possibility for the function of the region mapped by the DNase hypersensitive site?
For the next three questions, consider the following information about a protein called Gcn5p. [This problem is based on Brownell et al. (1996) Cell 84: 843-851.]
[1] Gcn5p is needed for activation of some, but not all, genes in yeast.
[2] Gcn5p does not bind with high affinity to any particular site on DNA.
[3] Gcn5p will interact with acidic transcriptional activators.
[4] When incubated with histones and the following substrates, Gcn5p will have the designated effects. A + in the column under "Effect" means that the histones move slower than unmodified histones on a polyacrylamide gel that separates on the basis of charge, with the histones moving toward the negatively charged electrode. A - means that the treatment has no effect on the histones. S-adenosylmethionine is a substrate for some methyl transfer reactions, and NADH is the substrate for ADPribosyl-transferases.
Mixture Effect
Gcn5p + histones -
Gcn5p + histones + ATP -
Gcn5p + histones + S-adenosylmethionine -
Gcn5p + histones + acetyl-coenzyme A +
Gcn5p + histones + NADH -
20.3 What conclusion is consistent with these observations?
20.4 What enzymatic activity is associated with Gcn5p?
20.5 Which step in the gene expression pathway is likely to be regulated by Gcn5p?
20.6 What functions have been ascribed to the locus control region of mammalian beta-globin genes?
20.7 Use the following information to answer the next 6 parts (a-f) of this question. The regulatory scheme is imaginary but illustrative of some of the models we have discussed.
The protein surfactin is produced in the lung to provide surface area for efficient gas exchange in the alveoli. Let's suppose that expression of the surfactin gene is induced in lung cells by a new polypeptide hormone called pulmonin. Induction by pulmonin requires a particular DNA sequence upstream of the surfactin gene; this is called PRE for pulmonin response element. Proteins that bind specifically to that site were isolated, and the most highly purified fraction that bound to the PRE contained two polypeptides. A cDNA clone was isolated that encoded one of the polypeptides called NFL2. Antisera that specifically recognizes NFL2 is available.
The mechanism of the induction by pulmonin was investigated by testing various cell fractions (nuclear or cytoplasmic) from uninduced or pulmonin‑induced lung cells in two assays. The presence or absence of NFL2 polypeptide was determined by reacting with the anti‑NFL2 antisera, and the ability to bind to the PRE DNA sequence was tested by an electrophoretic mobility shift assay. In a further series of experiments, the NFL2 polypeptide was synthesized in vitro by transcribing the cDNA clone and translating that artificial mRNA. The product has the same amino acid sequence as the native polypeptide and is referred to below as "expressed cDNA." The expressed cDNA (which is the polypeptide synthesized in vitro) was tested in the same assays, before and after treatment with the cytoplasmic and nuclear extracts and also with a protein kinase that will phosphorylate the expressed cDNA on a specific serine.
|
Line |
Source of protein
and Type of treatment |
React with anti‑NFL2 |
Bind to PRE DNA |
|
1 |
Uninduced cell cytoplasmic extract = unind. CE |
+ |
‑ |
|
2 |
Uninduced cell nuclear extract = unind. NE |
‑ |
‑ |
|
3 |
Induced cell cytoplasmic extract = ind. CE |
‑ |
‑ |
|
4 |
Induced cell nuclear extract = ind. NE |
+ |
+ |
|
5 |
Induced cell nuclear extract + phosphatase |
+ |
‑ |
|
6 |
Expressed cDNA |
+ |
‑ |
|
7 |
Expressed cDNA + ind. CE |
+ |
‑ |
|
8 |
Expressed cDNA + unind. NE |
+ |
‑ |
|
9 |
Expressed cDNA + ind. CE + unind. NE |
+ |
+ |
|
10 |
Expressed cDNA + unind. CE + unind. NE |
+ |
‑ |
|
11 |
Expressed cDNA + protein kinase + ATP |
+ |
‑ |
|
12 |
Expressed cDNA + protein kinase + ATP + unind. NE |
+ |
+ |
|
13 |
Expressed cDNA + protein kinase + ATP + ind. CE |
+ |
‑ |
Based on these data, an affinity column was made with the expressed NFL2 cDNA as the ligand and used to test binding of proteins from nuclear extracts. When the column was pretreated with protein kinase + ATP (so that NFL2 was phosphorylated), a ubiquitous nuclear protein called UBF3 was bound from nuclear extracts from both induced and uninduced cells. If the NFL2 ligand was not phosphorylated, no binding of nuclear proteins was observed.
To confirm that NFL2 really was part of the protein complex on PRE, antibodies against NFL2 were shown to react with this protein‑DNA complex. Furthermore, antibodies against phosphoserine, but not antibodies against phosphotyrosine, reacted with the specific PRE‑protein complex.
Answer questions a to f based on the above observations.
a) Where is the NFL2 polypeptide? (Use data in lines 1‑5.)
b) Where is the activity that will bind to the PRE site in DNA? (Use data in lines 1‑5.)
c) From the data in lines 6‑13, what must happen to the in vitro synthesized NFL2 (the expressed cDNA) in order to bind to the PRE site?
d) What proteins and covalent modifications of them are required to bind to the PRE site?
e) Which cell compartment has the protein kinase that acts on NFL2?
f) What model for pulmonin induction of the surfactin gene best fits the data given?