Multiple phenotypic features have been associated with 16p12.2
deletion (previously known as 16p12.1 deletion). Although not all
clinical features are completely recognizable in very young subjects,
most subjects show developmental delay and learning disability.
Affected individuals may present speech delay, hypotonia,
craniofacial and skeletal abnormalities, growth retardation,
microcephaly, cardiac disease. Furthermore, seizure disorders are
present and may manifest in several forms, including West syndrome,
febrile seizures or seizure-like episodes. Psychiatric and behavioral
abnormalities have also been documented. Individuals carrying 16p12.2
deletion do not present a common facial gestalt and do not share
identical clinical presentation, which indicates that this microdeletion
is nonsyndromic.
Most genomic disorders are the result of a non-allelic homologous
recombination (NAHR) between large (larger than 10kb) and highly similar
sequences, called segmental duplications. Several chromosomes are
enriched in these types of DNA sequences, such as chromosome 16, most
particularly its short arm. This leads to a higher frequency of this
rearrangements and therefore, genomic disorders associated with this
deletion/duplication.
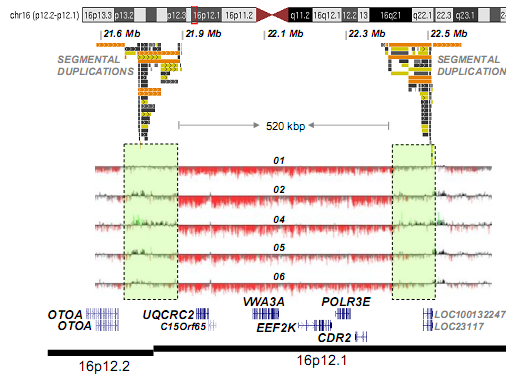
16p12.2 microdeletion expands approximately 500kb, leading to a
hemizygous state of six genes: UQCRC, C15orf65, VWA3A, EEF2K, POLR3E,
CDR2 (Figure right). 16p12.2 microdeletion can be diagnosed by
Fluorescent In Situ Hybridization (FISH) and Array Comparative Genomic
Hybridization (array CGH). This molecular alteration cannot be
identified by routine analysis of G-banded chromosomes or other
conventional cytogenetic techniques
Assessing family history is essential in 16p12.2 microdeletion. In
stark contrast to other syndromic disorders where most CNVs are de novo,
this deletion was inherited from a parent in 95% of the cases.
